Watching molecules wobble
We are interested in the structure and motion of biomolecules, and how these relate to their functional and evolutionary roles in biology. We do everything from developing techniques for in silico investigation of biomolecular structure and interactions, to applying these in diverse areas ranging from drug development to fathoming the earliest stages of protein evolution. While much of our research is at a fundamental level, we’re not averse to working on things that might be useful in the near(ish) future. The things we’re most excited about at the moment include:
Anti-microbial peptide specificity and mode of action
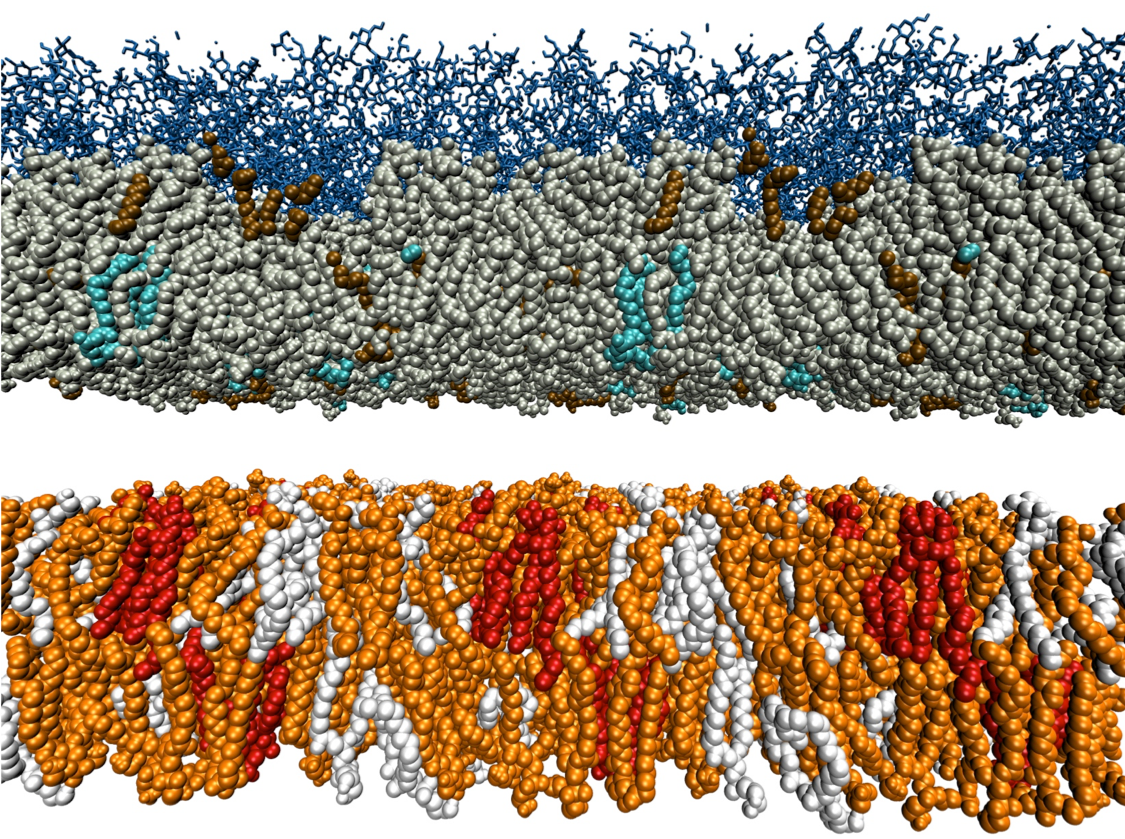 Anti-microbial resistance is a growing problem, with new treatments urgently needed. Naturally-occurring antimicrobial peptides (AMPs) are the tools many living organisms employ to defend themselves against bacterial attack. We are part of the Maurice Wilkins Centre Flagship Programme “Tackling Antimicrobial Resistance with Antimicrobial Peptides”, lead by Dist. Prof. Margaret Brimble and Prof. Greg Cook. We are developing realistic models for the cell membranes of a range of different bacterial species, and making use of our automated parameterisation software to rapidly build models for peptide variants, and then testing their efficacy and mode of action using MD simulations. Our results will help guide the efforts of synthetic chemists and cell biologists who will characterise the peptides experimentally.
Anti-microbial resistance is a growing problem, with new treatments urgently needed. Naturally-occurring antimicrobial peptides (AMPs) are the tools many living organisms employ to defend themselves against bacterial attack. We are part of the Maurice Wilkins Centre Flagship Programme “Tackling Antimicrobial Resistance with Antimicrobial Peptides”, lead by Dist. Prof. Margaret Brimble and Prof. Greg Cook. We are developing realistic models for the cell membranes of a range of different bacterial species, and making use of our automated parameterisation software to rapidly build models for peptide variants, and then testing their efficacy and mode of action using MD simulations. Our results will help guide the efforts of synthetic chemists and cell biologists who will characterise the peptides experimentally.
Where proteins and membranes meet
 Characterisation of protein-membrane interactions is vital due to their central importance in cellular metabolism and signal transduction, yet they are notoriously demanding to investigate experimentally due to the difficulty in including lipid phases in most techniques for studying protein structure and function. We have a number of ongoing projects involving the use of molecular dynamics simulations to probe protein- or peptide-membrane interactions for a range of different systems, including the novel cancer therapeutic target phosphoinositide 3-kinase (PI3Kα, with Dr Jack Flanagan, Auckland Cancer Research Institute and Prof. Peter Shepherd, University of Auckland); membrane-penetrating anti-microbial and anti-cancer peptides (with Viji Sarojini, University of Auckland), and the anti-fungal cytochrome P450 (with Assoc. Prof. Joel Tyndall, University of Otago).
Characterisation of protein-membrane interactions is vital due to their central importance in cellular metabolism and signal transduction, yet they are notoriously demanding to investigate experimentally due to the difficulty in including lipid phases in most techniques for studying protein structure and function. We have a number of ongoing projects involving the use of molecular dynamics simulations to probe protein- or peptide-membrane interactions for a range of different systems, including the novel cancer therapeutic target phosphoinositide 3-kinase (PI3Kα, with Dr Jack Flanagan, Auckland Cancer Research Institute and Prof. Peter Shepherd, University of Auckland); membrane-penetrating anti-microbial and anti-cancer peptides (with Viji Sarojini, University of Auckland), and the anti-fungal cytochrome P450 (with Assoc. Prof. Joel Tyndall, University of Otago).
Structural phylogenetics
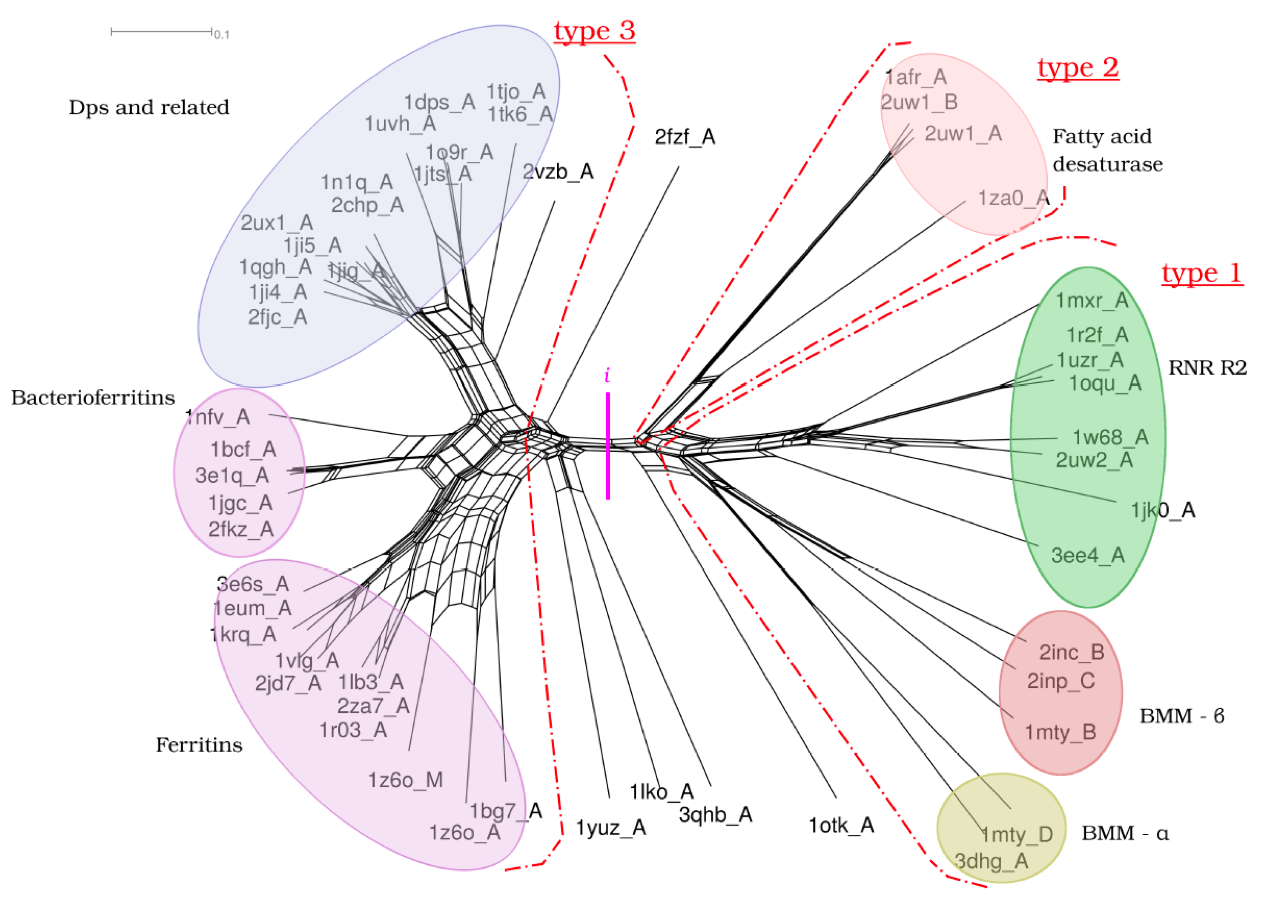 We are working with Prof. Ant Poole, University of Auckland, on applying molecular dynamics simulations to evolutionary biology to probe deep evolutionary relationships and to recategorise the protein structure universe. Sophisticated phylogenetic methods have been developed to exploit the avalanche of sequence data currently being generated, but these fail at long evolutionary distances, as the similarities become too low – the so-called “twilight zone”. We are taking advantage of the fact that protein structure is better conserved than sequence, along with the recent exponential growth of structural information for extant species and the capacity of molecular dynamics simulations to explore structural flexibility to provide new ways of examining relationships between protein structures and of studying the earliest events in the history of life, where evolutionary signal is scant.
We are working with Prof. Ant Poole, University of Auckland, on applying molecular dynamics simulations to evolutionary biology to probe deep evolutionary relationships and to recategorise the protein structure universe. Sophisticated phylogenetic methods have been developed to exploit the avalanche of sequence data currently being generated, but these fail at long evolutionary distances, as the similarities become too low – the so-called “twilight zone”. We are taking advantage of the fact that protein structure is better conserved than sequence, along with the recent exponential growth of structural information for extant species and the capacity of molecular dynamics simulations to explore structural flexibility to provide new ways of examining relationships between protein structures and of studying the earliest events in the history of life, where evolutionary signal is scant.
Host-guest binding of fluctional molecules
 Bullvalenes are shapeshifting molecules that spontaneously isomerase in solution. We are working with Dr Thomas Fallon, Massey University to characterise how bullvalenes bind to host cyclodextrin molecules, including whether host binding is isomer-specific and can provide a way to trap particular isomers. We have developed new rapid screening procedures for identifying potential binding modes, and validating our results against NMR data.
Bullvalenes are shapeshifting molecules that spontaneously isomerase in solution. We are working with Dr Thomas Fallon, Massey University to characterise how bullvalenes bind to host cyclodextrin molecules, including whether host binding is isomer-specific and can provide a way to trap particular isomers. We have developed new rapid screening procedures for identifying potential binding modes, and validating our results against NMR data.
Automated parameterisation of atomic-level models
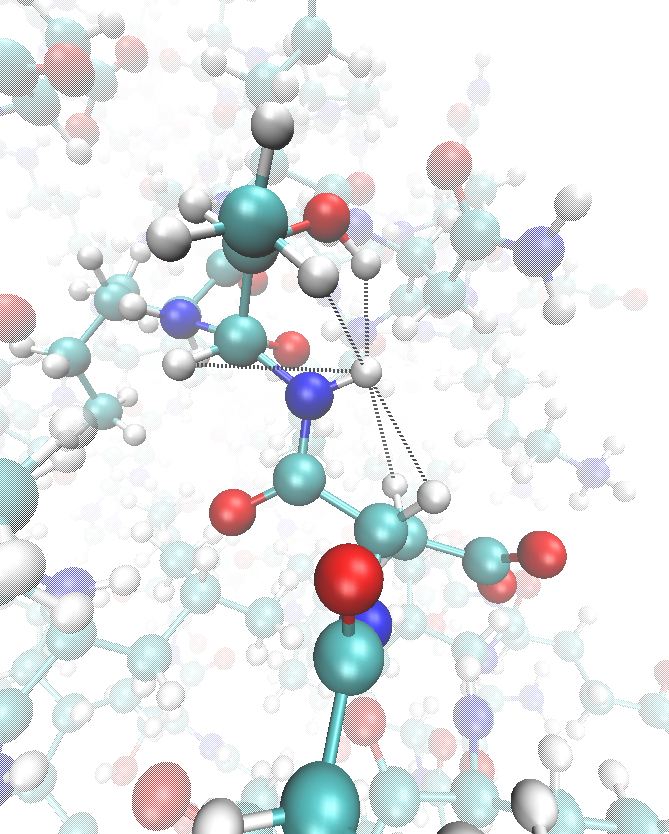 While molecular dynamics simulations are a powerful way to probe molecular structure and motion at an atomic level of detail, they have traditionally been highly reductionist, with a typical atomic-level simulation system comprising a single protein molecule surrounded by water – a far cry from cellular conditions. One reason for this austerity is that reliable parameter sets are only available for the most common biomolecules, and manual parameterisation of new molecules is error-prone, tedious and slow. We are working with the group of Alan Mark (University of Queensland) to develop a high-throughput framework for automated parameter assignment and optimisation that will generate reliable parameter sets for almost any biomolecule imaginable.
While molecular dynamics simulations are a powerful way to probe molecular structure and motion at an atomic level of detail, they have traditionally been highly reductionist, with a typical atomic-level simulation system comprising a single protein molecule surrounded by water – a far cry from cellular conditions. One reason for this austerity is that reliable parameter sets are only available for the most common biomolecules, and manual parameterisation of new molecules is error-prone, tedious and slow. We are working with the group of Alan Mark (University of Queensland) to develop a high-throughput framework for automated parameter assignment and optimisation that will generate reliable parameter sets for almost any biomolecule imaginable.
Coupling NMR data with MD simulations
 As well as using NMR data, when available, to validate or bias our simulations, we work on development of methodology for combining NMR data with molecular dynamics simulations. Lukas Wirz, a former PhD student, developed and implemented of novel means of biasing molecular dynamics simulations to fit experimentally measured NMR residual dipolar couplings into the GROMOS biomolecular simulation software, for which we form part of the development team. We also have an ongoing collaboration with Assist. Prof. Lorna Smith (University of Oxford) in this area.
As well as using NMR data, when available, to validate or bias our simulations, we work on development of methodology for combining NMR data with molecular dynamics simulations. Lukas Wirz, a former PhD student, developed and implemented of novel means of biasing molecular dynamics simulations to fit experimentally measured NMR residual dipolar couplings into the GROMOS biomolecular simulation software, for which we form part of the development team. We also have an ongoing collaboration with Assist. Prof. Lorna Smith (University of Oxford) in this area.
Development of supra-atomic lipid models for multi-scale simulation
 To allow exploration of the behaviour of biological systems larger spatial and temporal scales, we are developing supra-atomic (“coarse-grained”) lipid models together with Assist. Prof. Sereina Riniker and Emiritus Prof. Wilfred van Gunsteren (ETH Zurich). These models will be compatible with the GROMOS supra-molecular solvent models and atomic-level solvent and biomolecule models, enabling multi-scale simulation of large, complex biological systems for biologically relevant timescales. Additionally, because they will be polarisable, they will be responsive to the local electronic environment in a way that many coarse-grained models are not.
To allow exploration of the behaviour of biological systems larger spatial and temporal scales, we are developing supra-atomic (“coarse-grained”) lipid models together with Assist. Prof. Sereina Riniker and Emiritus Prof. Wilfred van Gunsteren (ETH Zurich). These models will be compatible with the GROMOS supra-molecular solvent models and atomic-level solvent and biomolecule models, enabling multi-scale simulation of large, complex biological systems for biologically relevant timescales. Additionally, because they will be polarisable, they will be responsive to the local electronic environment in a way that many coarse-grained models are not.